Diagnose
Morbus Pompe, was ist das?
Ich möchte hier zunächst eine einfache und allgemein verständliche Information zu diesem Thema geben.
Es handelt sich bei dieser Muskelerkrankung um eine genetisch bedingte und vererbbare Stoffwechselstörung. Die genaue Bezeichnung lautet: Myopathie bei Mangel der sauren Maltase (1,4 alpha-Glucosidase-Mangel; Glykogenose Typ II; Morbus-Pompe).
Andere Bezeichnungen (Synonyme) für diese Erkrankung lauten:
Pompe Krankheit, Glykogenspeicherkrankheit Typ II, Glykogenose Typ II, Saure Maltase Mangel
Insgesamt gibt es 3 bekannte und in der Literatur beschriebene Verlaufsformen der Glykogenose Typ II „Pompe-Krankheit:“
- Säuglings- bzw. Kindheitsform (infantiler Typ II a)
- Jugendlichenform (juveniler Typ II b )
- Erwachsenenform (adulter Typ II c).
In neuerer Zeit unterscheidet man auch zwischen „early onset“ und „late onset“.
Eine ausführliche Darstellung der verschiedenen Verlaufsformen findet sich auch auf dieser Website: http://www.mpompe.de/pompe/verlaufsformen
Was passiert nun bei dieser Erkrankung?
In den Muskelzellen wird Glykogen (i.w.S. Zucker) eingelagert. Im Normalfall, also bei einem „Gesunden“ wird das Glykogen mittels verschiedener Enzyme aus der Zelle herausgelöst und in Energie (körperliche Kraft) umgewandelt. Eines dieser Enzyme ist die Saure Maltase. Bei Morbus Pompe handelt es sich um einen „Mangel an saurer Maltase“ mit dem Ergebnis, dass sich Teile des Glykogen nicht aus den Muskelzellen (bei Morbus Pompe speziell aus den Lysosomen) herauslösen. Die Zellen speichern und speichern und gehen letztendlich zu Grunde.
This file is licensed under the Creative Commons Attribution ShareAlike 2.5, Attribution ShareAlike 2.0 and Attribution ShareAlike 1.0 License.
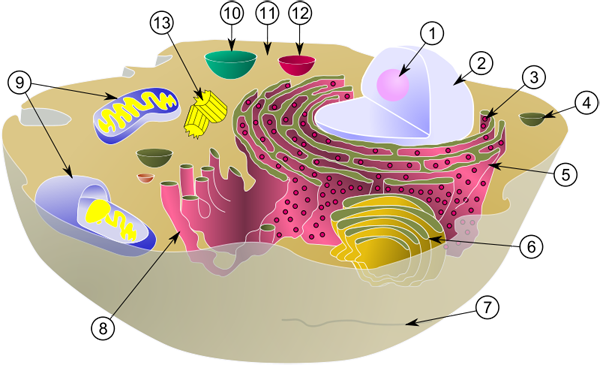
1. Nukleolus. 2. Zellkern (Nukleus). 3. Ribosomen. 4. Vesikel. 5. Rauhes Endoplasmatisches Reticulum (ER). 6. Golgi-Apparat. 7. Mikrotubuli. 8. Glattes ER. 9. Mitochondrien. 10. Vakuole. 11. Zytoplasma. 12. Lysosom. 13. Zentriolen. (Quelle: https://de.wikipedia.org/wiki/Zelle_(Biologie))
Wie merkt man es? – Symptome –
Man stellt an sich selbst folgende Symptome (Hauptmerkmale) fest:
- allgemeiner Kräfteverfalll,
- körperliche Schwäche, besonders in den Oberschenkeln, der Rückenmuskulatur und des Schultergürtelbereiches,
- Kraftlosigkeit, keine Ausdauer mehr,
- unsicherer Gang (Watschel- oder Enten-Gang) mit Neigung zum stolpern und zu Stürzen
- ständige Müdigkeit und Mattheit,
- Kurzatmigkeit,
- einfache Dinge wie Treppen steigen, Fahrrad fahren oder größere Gegenstände heben, fallen deutlich schwerer als „normalerweise“.
Wenn man den Kräftemangel selbst deutlich bemerkt, ist bereits ca. 40 % der Muskelmasse verloren. Dies ist eine Zirka-Messgröße. Die Angaben schwanken in der einschlägigen Literatur zwischen 30% und 50%.
Teilweise können noch folgende Symptome hinzukommen*:
- Ödeme
- Konzentrationsstörungen
- Wortfindungsstörungen
- Herzklopfen
- Abgeschlagenheit
- Nervosität
- Tachykardie
- Tremor
- Herzrhythmusstörungen
- Depression
- Infektionen
- Angstzustände
- Sekretretention (die Rückhaltung von Sekret)
- Einschlafneigung
- Cyanose
- Schlafstörungen
- Dyspnoe
- Alpträume
- Tachypnoe
- Kopfschmerzen
- Orthopnoe (Zustand höchster Atemnot, in dem nur bei aufgerichtetem Oberkörper genügend Atemluft in die Lunge gelangt)
- Nackenschmerzen
- Schwindel
- Einsatz der Atemhilfsmuskeln
Wenn einige von diesen Symptome zu den oben genannten schon dazugekommen sind, ist möglicherweise bereits auch die Atmungsmuskulatur betroffen!
*Quelle: Prof. Dr. Laier-Groeneveld, Vortrag: „Nichtinvasive und invasive Beatmung bei Kindern und Erwachsenen“, Beatmungsworkshop Pompe Deutschland e.V. vom 06.10.2018 in Essen
Wie wird die Erkrankung diagnostiziert?
In Kombination mit den persönlichen Symptomen (s.o.) ist ein erster Untersuchungsansatz, um die Frage zu klären, ob diese Erkrankung bei einem selbst vorliegt, die Blutuntersuchung beim Arzt. Es ist hier besonders der Arzt des persönlichen Vertrauens zu wählen, denn dieser wird sich zukünftig intensiver mit dieser Thematik beschäftigen müssen. Bei Morbus Pompe sind i.d.R. einige spezifische Leberwerte zu hoch. Klassischer Weise sind zunächst erst mal die sogenannten Muskelenzymwerte (CK) (Kreatinkinase = sogenannte Serumenzyme) und die LDH-Werte (Lactatdehydrogenase) erhöht. Ein erhöhter CK-Wert wird standardmäßig von den Ärzten zunächst als Indiz für einen Herzinfarkt gewertet. Um diese Diagnose „Herzinfarkt“ auszuschließen, ist hier der spezielle CK-mb Wert zu untersuchen. Aber auch andere spezielle Leberwerte wie GOT (Glutamat- Oxalazetat- Transaminase) und GPT (Glutamat- Pyuvat- Transaminase) oder aber auch Aldolase und Harnsäure können erhöht sein.
Der nächste Schritt ist dann, natürlich immer in Absprache mit dem behandelnden Arzt, eine Untersuchung beim Neurologen. Hier wird ein EMG (Elektro-Myo-Graphie) gemacht. Fällt dies ebenfalls ungünstig aus und lassen sich die vielen anderen Muskelerkrankungen (Dystrophien und Atrophien) ausschließen, so kann der nächste Untersuchungsschritt erfolgen.
Hier gibt es nun verschiedene diagnostische Verfahren:
- Trockenbluttest
- MRT
- Muskelbiopsie / Hautbiopsie
- Genetische Untersuchung
Trockenbluttest
Seit einiger Zeit ist die Diagnostik mittels eines sogenannten Trockenbluttests (Fachbegriff: Dried blood spot) möglich. Dieses Verfahren erlaubt eine zuverlässige Diagnose eines Morbus Pompe aus nur wenigen Tropfen Blut, die auf eine spezielle Filterkarte getropft und in einem Speziallabor analysiert werden.
Der Trockenbluttest kann in diesem Labor durchgeführt werden: Stoffwechsellabor der Klinik und Poliklinik für Kinder- und Jugendmedizin am Universitätsklinikum Hamburg-Eppendorf (UKE).
MRT
Die Magnetresonanztomographie (MRT) kann als ergänzendes diagnostisches Verfahren angewendet werden. Hierbei kann man erkennen wieviel Muskelmasse (stellt sich im MRT dunkel dar) bereits in Fett-/Bindegewebe umgewandelt wurde (im MRT weiß dargestellt). Ein durchaus geeignetes Verfahren, um den Verlauf der Erkrankung in regelmäßigen Abständen optisch zu kontrollieren.
Muskelbiopsie / Hautbiopsie
Objektiv nachweisen lässt sich der „Saure Maltase Mangel“ auch, wenn eine Muskelbiopsie durchgeführt wird. Es wird aus einem definierten Bereich (meist Oberschenkel) ein kleiner Muskelfaser-Strang herausoperiert. Alternativ kann auch eine Hautbiopsie durchgeführt werden.
Bei der Muskelbiopsie ist es wichtig, dass die zu untersuchenden Muskelfaserzellen nicht zerstört werden. Deshalb sollte man bei der Wahl des Operateurs sorgfältig vorgehen und nicht zum erstbesten Chirurgen gehen, der zwar schnell schneidet, aber vielleicht die Wichtigkeit der Unversehrtheit der entnommenen Probe nicht erkennt. Die Zellen können beim Schneiden so zerstört werden, dass eine sorgfältige Diagnose nicht mehr möglich ist. Da die Muskelentnahme ohne Voll-Narkose (nur unter örtlicher Betäubung) gemacht wird und etwas unangenehm ist, sollte man einen zweiten Versuch nicht durch die Wahl eines für diese Zwecke ungeeigneten Chirurgen provozieren.
Die entnommene Muskelprobe wird nun histochemisch untersucht. Hier lässt sich nun ziemlich genau die Art der Glykogenose bestimmen und, bei genügend Muskelmasse, auch eine möglicherweise vorhandene Unterart (Mutation) der Erkrankung. Auf jeden Fall sollte man versuchen sicherzustellen, dass die Probe in geeigneter Weise aufbewahrt wird, um spätere Untersuchungen zu ermöglichen. Eine Mutationsanalyse kann in dafür geeigneten Kliniken bzw. Muskel-Zentren durchgeführt werden.
In Deutschland gibt es für diese Art Eingriff sicherlich einige gute Kliniken. Zu empfehlen sind hier aber Universitätskliniken, die ein entsprechendes Muskellabor oder zumindest die geeigneten Untersuchungslabors vorzuweisen haben, damit die Probe ohne große Transportwege untersucht werden kann.
Da die Muskelbiopsie von den Patienten als äußerst unangenehm empfunden wird und es auch andere geeignete, weniger invasive Diagnoseverfahren gibt, gehört die Muskelbiopsie heute nicht mehr unbedingt zu den Untersuchungsmethoden der ersten Wahl.
Genetische Untersuchung
Die sicherste Diagnosemethode ist die genetische Untersuchung. Die genetische Diagnostik hat sich in den letzten Jahren deutlich verbessert. Musste man vor einigen Jahren noch Monate bis Jahre auf die das Ergebnis warten, liegt das Ergebnis heute bereits innnerhalb kurzer Zeit (Erfahrungswert: i.d.R. 4-6 Wochen) vor. Bei der genetischen Untersuchung wird auch die GAA-Mutation festgestellt. Die Art der Mutation kann zusammen mit den anderen diagnostischen Ergebnissen (Blutwerte, Restenzmyaktivität,etc.) – man muss hier das „Gesamtpaket“ sehen – eventuell gewisse Rückschlüsse auf den Verlauf der Erkrankung geben. Eine umfassende genetische Beratung ist hier empfehlenswert.